Hội chứng Angelman (angelman sydrome, viết tắt
AS) là một
rối loạn di truyền, chủ yếu ảnh hưởng đến
hệ thống thần kinh.
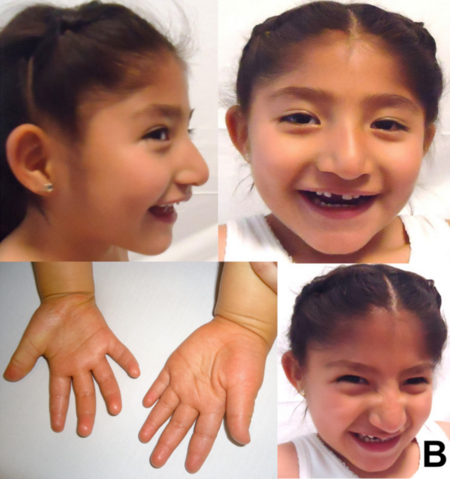
[2] Các triệu chứng bao gồm một đầu nhỏ và một diện mạo đặc thù,
khuyết tật trí tuệ nghiêm trọng,
khuyết tật phát triển, vấn đề về nói, cân bằng và các vấn đề vận động, hay co giật và khó ngủ.
[2] Trẻ em thường có tính cách vui vẻ và có hứng thú đặc biệt với nước.
[2] Các triệu chứng thường trở nên đáng chú ý từ một tuổi trở đi.
[2]Hội chứng Angelman thường là do
đột biến mới chứ không phải do
di truyền từ cha mẹ của một người.
[2] Hội chứng Angelman là do thiếu chức năng của một phần
nhiễm sắc thể 15 được thừa hưởng từ mẹ của một người.
[2] Hầu hết thời gian, đó là do việc xóa hoặc đột biến gen
UBE3A trên nhiễm sắc thể đó.
[2] Đôi khi, đó là do thừa hưởng hai bản sao nhiễm sắc thể 15 từ cha của một người và không có từ mẹ của họ.
[2] Vì các phiên bản của người cha bị bất hoạt bởi một quá trình được gọi là in dấu gen, nên không còn phiên bản chức năng của gen.
[2] Chẩn đoán dựa trên các triệu chứng và có thể là
xét nghiệm di truyền.
[3]Hiện tại không có cách chữa trị nào.
[3] Điều trị nói chung là hỗ trợ trong môi trường tự nhiên.
[3] Thuốc chống động kinh được sử dụng ở những người bị động kinh.
[3] Vật lý trị liệu và dây kẹp có thể giúp bệnh nhân đi bộ.
[3] Những người bị ảnh hưởng có
tuổi thọ trung bình gần như bình thường.
[2]Hội chứng Angelman ảnh hưởng đến 1 trên 12.000 đến 20.000 người.
[2] Nam và nữ bị ảnh hưởng với tần suất bằng nhau.
[3] Nó được đặt theo tên của một
bác sĩ nhi khoa người Anh,
Harry Angelman, người đầu tiên mô tả
hội chứng này vào năm 1965.
[3][4] Một thuật ngữ cũ hơn của nó, "hội chứng rối vui vẻ", thường được coi là mang tính miệt thị.
[5] Hội chứng Prader-Willi là một tình trạng riêng biệt, gây ra bởi sự mất tương tự nhiễm sắc thể 15 của người cha.
[6]